
By James O’Brien
Immunotherapy is emerging as the fourth pillar of cancer therapy alongside traditional surgery, chemotherapy and radiotherapy treatments. By harnessing the anti-cancer properties of immune cells, immunotherapy has had particular success in targeting blood cancers. Nevertheless, various limitations have restricted immunotherapy’s broader application in cancer therapy, particularly in treating solid tumours. The recent discovery of a unique tumour-resident innate-like T-cell population, and their proposed application in immunotherapy, may help address these issues.
Immunotherapy describes the modulation of immune cells to enhance their anti-cancer effects. This can range from engineering cytotoxic (‘killer’) T-cells to target specific cancers through the use of tumour-specific chimeric antigen receptors (CARs), to drugs inhibiting immune checkpoints: molecular mechanisms that dampen anti-cancer immune responses. Although these therapeutics have shown promise, several limitations currently exist. For example, anti-cancer T-cells can become ‘exhausted’ after prolonged activity, penetration of T-cells into solid tumours is difficult, and knowledge of tumour-specific, non-self antigens (neoantigens) is needed. In addition, many patients do not respond to immune checkpoint blockade therapies.
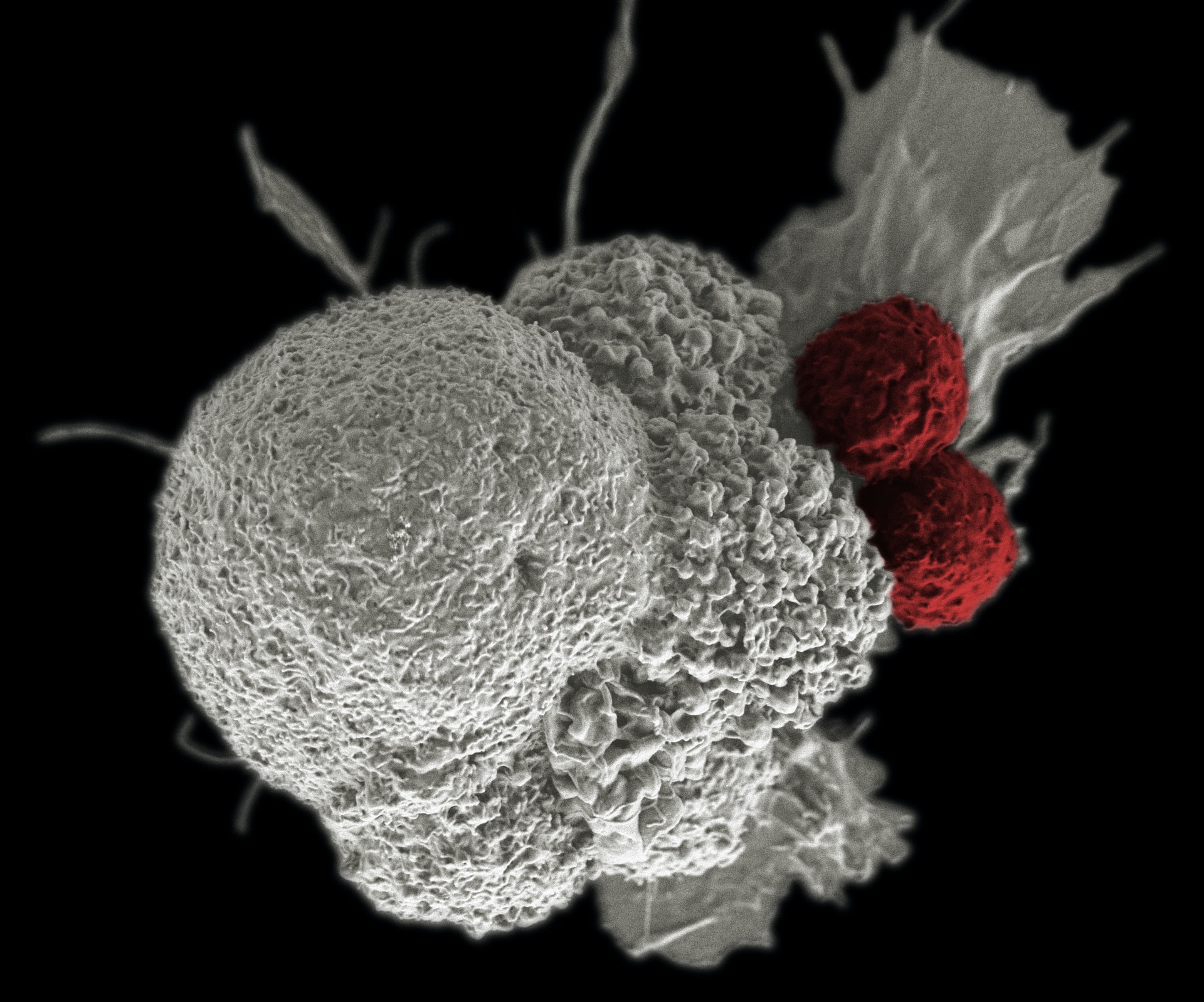
By harnessing the anti-cancer properties of immune cells, immunotherapy has had particular success in targeting blood cancers.
A recent survey of T-cells resident in mouse and human tumours identified a unique killer T- cell population that could overcome these limitations. These innate-like killer T-cells (ILTCKs) differed in several ways to the killer T-cells currently used in CAR therapies. ILTCKs were found to downregulate PD-1, a key immune checkpoint receptor, so did not become ‘exhausted’ after prolonged activity. The absence of immune checkpoint regulation would also make ILTCKs useful in treating tumours that do not respond to immune checkpoint blockade therapies. ILTCKs were also found to penetrate more deeply into solid tumours than other T-cell populations, suggesting enhanced tumour infiltration properties and survival in the hostile tumour microenvironment. Moreover, unlike the T-cells used in CAR therapies, ILTCKs did not depend on antigen presenting cells (APCs) for activation. Instead, ILTCKs were constitutively primed when resident in tumours. Together, these features make ILTCKs an attractive option for future T-cell based immunotherapies.
Interestingly, ILTCKs were also found to recognise self-antigens on tumours, in contrast to other tumour-resident T-cell populations that recognise tumour-specific, non-self antigens (neoantigens). Current CAR T-cell therapies utilise tumour-resident T cells, meaning knowledge of a tumour’s specific neoantigens is required. Use of ILTCKs in future T-cell based therapies would remove this requirement, leading to faster treatment administration and easier application to a wider range of tumours.
Nonetheless, ILTCK’s recognition of self-antigens raised the question of how ILTCKs do not elicit autoimmunity–where the body’s immune system attacks its own tissues. Normally, developing T-cells that react with self-antigens are eliminated in the thymus to prevent autoimmunity. It was found, however, that ILTCKs escape this fate. Instead, ILTCK T-cell receptor (TCR) machinery was dampened down and activating receptors were downregulated, rendering ILTCKs harmless to the body’s own tissues. This finding raised the additional question of how ILTCKs are stimulated to specifically attack cancer cells.
ILTCKs are a unique population of anti-cancer T-cells that do not appear to have many of the limitations faced by current immunotherapies.
The answer came in the discovery that IL-15, a signalling molecule produced at high levels by cancer cells but not normal body cells, stimulates ILTCK effector differentiation. This process involves upregulation of activating receptors, increased sensitivity of TCR machinery and downregulation of the immune checkpoint molecule PD-1, allowing ILTCKs to kill cancer cells effectively. This essential role of IL-15 was highlighted by experiments that showed deletion of IL-15 from cancer cells abrogated protection provided by ILTCKs, leading to increased tumour growth.
IL-15’s crucial role in ILTCK effector differentiation led to attempts to modulate the induction of IL-15 signalling in ILTCKs. This involved engineering of a drug-inducible, constitutively active form of the transcription factor STAT5B (STAT5B-CA) into ILTCKs to coordinates the transcription programme downstream of IL-15 signalling. Transfer of these engineered ILTCKs into tumour-prone mice resulted in ILTCKs rapidly colonising tumours and tumour growth being reduced. These initial results suggest that modulating IL-15 signalling could be a potent tool in future ILTCK-based immunotherapies.
ILTCKs are a unique population of anti-cancer T-cells that do not appear to have many of the limitations faced by current immunotherapies. In addition, IL-15 signalling has been successfully modulated in ILTCKs, representing a first step in ILTCK engineering for cancer therapy. However, translating these exciting results into effective therapeutics may not be straightforward. Although ILTCKs were found in human malignancies, the majority of ILTCK characterisation and associated experiments used mouse samples. It remains to be seen whether these findings will be replicated with human-derived ILTCKs, with initial indications of more complex immune signalling in human ILTCKs. Nonetheless, ILTCKs have shown the potential to become a potent weapon in future immunotherapy arsenals.
This article is based on the paper: Chou, C., Zhang, X., Krishna, C., Nixon, B. G., Dadi, S., Capistrano, K. J., Kansler, E. R., Steele, M., Han, J., Shyu, A., Zhang, J., Stamatiades, E. G., Liu, M., Li, S., Do, M. H., Edwards, C., Kang, D. S., Chen, C. T., Wei, I. H., Pappou, E. P., … Li, M. O. (2022). Programme of self-reactive innate-like T cell-mediated cancer immunity. Nature, 605(7908), 139–145. https://doi.org/10.1038/s41586-022-04632-1



