By Rhian Gruar
This article was originally published in The Oxford Scientist Michaelmas Term 2021 edition, Change.
On the 26 June 2000, President Bill Clinton announced the completion of the first draft of the Human Genome Project (HGP) to the world, ushering in a new age of scientific understanding. The HGP was a decade-long endeavour to decode the sequence of our DNA and promised to revolutionise the world. In the following years, doubts have been cast as to whether this project was as momentous as initially presented. Merely knowing the sequence of a gene doesn’t immediately expose its function, to say nothing of the 98% of our genome that is absent of genes.
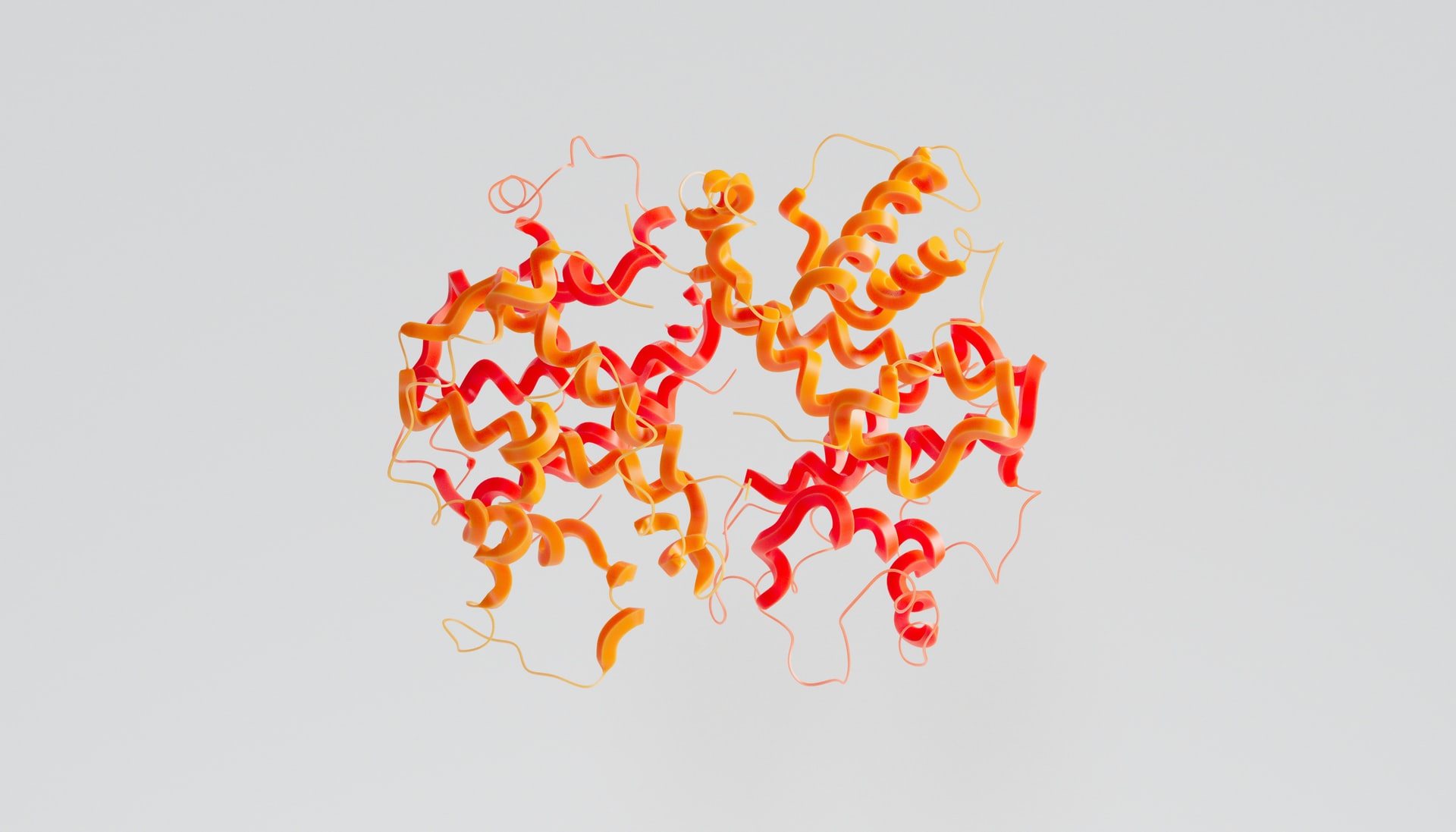
Over two decades later in November 2020, Google’s DeepMind team and their collaborators at the European Bioinformatics Institute announced the development of AlphaFold2, an artificial intelligence (AI) tool capable of predicting the 3D structure of a protein from its sequence alone. A follow up announcement in July 2021 released the source code for the tool, alongside a publicly accessible database containing predicted structures of the entire human proteome, the complete set of proteins expressed by a human (which is about 20,000 proteins).
Merely knowing the sequence of a gene doesn’t immediately expose its function, to say nothing of the 98% of our genome that is absent of genes.
Scientists such as Professor Venki Ramakrishnan, Nobel laureate and president of the Royal Society, have heralded AlphaFold2 as a ‘stunning advance [towards solving] a 50-year-old grand challenge in biology’ and something that will ‘fundamentally change biological research’. This ‘grand challenge’ is the protein folding problem, and it has been baffling scientists for years. How do proteins go from a linear sequence of amino acids to a 3D folded protein?
According to the Leventhal Paradox, it would take longer than the known age of the universe for an amino acid to sample every possible configuration to find the most stable structure, yet a protein can fold in milliseconds. This is a very important process to understand as a protein’s structure is central to defining its function. For instance, collagen gains its elasticity from its fibrous structure which allows it to withstand high forces, and haemoglobin is only able to cooperatively bind oxygen and deliver it around the body due to the structure of its four subunits.
Therefore, to understand the function of a protein, it is paramount to know its structure. Historically, researchers have had to devote significant amounts of time to solve protein structures. Techniques such as X-ray crystallography and cryogenic electron microscopy (cryoEM), alongside having a high cost attached, require a long optimisation process to find the ideal conditions to solve a given protein’s structure. Often, these techniques—especially X-ray crystallography—don’t even result in a structure that is indicative of the protein’s actual structure in the cell.
Scientists such as Professor Venki Ramakrishnan, Nobel laureate and president of the Royal Society, have heralded AlphaFold2 as a ‘stunning advance [towards solving] a 50-year-old grand challenge in biology’ and something that will ‘fundamentally change biological research’.
DeepMind propose AlphaFold2 as the solution to all these issues. The AI tool has been trained on thousands of previously solved proteins structures to recognise patterns in the 3D structure of the given protein in relation to its linear sequence. Once given a sequence, AlphaFold2 can propose a possible structure in a matter of seconds—a vast improvement from experimental techniques that can sometimes take years to result in a structure. AlphaFold2 also provides a score of confidence in the predictions for the protein: 36% of proteins in their database are so precise that they contain atomic details of the protein structure, the gold standard for structural biologists. This is precise enough to be used for drug design.
In fact, research has already begun to benefit from AlphaFold2. A research group collaborating with DeepMind have used the tool to predict the structure of a SARS-Cov-2 spike protein. This structure was later experimentally validated. Some other groups are proposing using AlphaFold2 to aid in engineering enzymes to recycle single-use plastics.
Despite these successes, AlphaFold2 is not infallible, and it has some blind spots. Proteins with intrinsically disordered regions (areas that are highly flexible and do not settle into a defined structure) are less accurately predicted. AlphaFold2 is also limited to monomeric proteins, whose structures are not defined by their interactions with other proteins or biomolecules. This is a major short falling as multi-protein complexes control many processes central to life, and yet are some of the hardest structures to solve experimentally. Similarly, proteins that switch between multiple structures to carry out their function may be predicted by AlphaFold2 to have a structure that is intermediate of their true structures. Membrane proteins, proteins that are part of or interact with cell membranes, are also poorly predicted by AlphaFold due to the lack of existing solved structures of it to be trained on.
AlphaFold2 is also limited to monomeric proteins, whose structures are not defined by their interactions with other proteins or biomolecules. This is a major short falling as multi-protein complexes control many processes central to life.
These limitations are the same limitations that are experienced when solving structures experimentally. As such, structural biologists may have to hold off their panic about AI taking over their jobs. AlphaFold2 may have merely shortened the time frame for structural studies rather than expanding the horizon of possible structures. It will also be a long time before a structure will be accepted based solely off computational predictions—experimental validation is still required. Scientists should be prepared to collaborate with this new tool; AlphaFold2 predictions may be a useful starting point when processing X-ray crystallography data or a reference for interpreting cryoEM maps. It’s been years since structural biology was just a brute force trying to solve the structures of as many protein domains as possible. Currently, many researchers focus on interpreting structural information to give insight on dynamic functions, often using molecular dynamic simulations to perturb the environment and probe function.
So, AlphaFold2 has not solved the protein folding enigma, it merely proposes an alternative route to get to the final destination without providing insight on the route taken. Nonetheless, it is a useful tool available to scientists. Protein prediction is still in its nascent years and, much like the Human Genome Project before it, only time will tell if, and how, it will redefine structural biology.